
DNA sequenceの結果から、遺伝子変異を分かりやすい図にまとめる方法として、Waterfall plotがあります。Rでどのように描くか、実際にやりながら学んでみましょう。
はじめに
複数の腫瘍検体のsequenceを行った際に、遺伝子変異のデータを分かりやすい図で表現したい場合があります。論文でも以下のような図をよく見ると思います(oncoprintあるいはwaterfall plotと呼ばれます)。TCGAやICGCのパブリックデータベースを扱っているcBioPortalでは、WebツールOncoprinterが公開されており、自分の実験データでもアップロードするとwaterfall plotを綺麗に書いてくれます。
ただ実際に論文に使用する図は、細かい調整が必要です。RでGenVisRパッケージを使うことで、以下のような図を作成することができます。
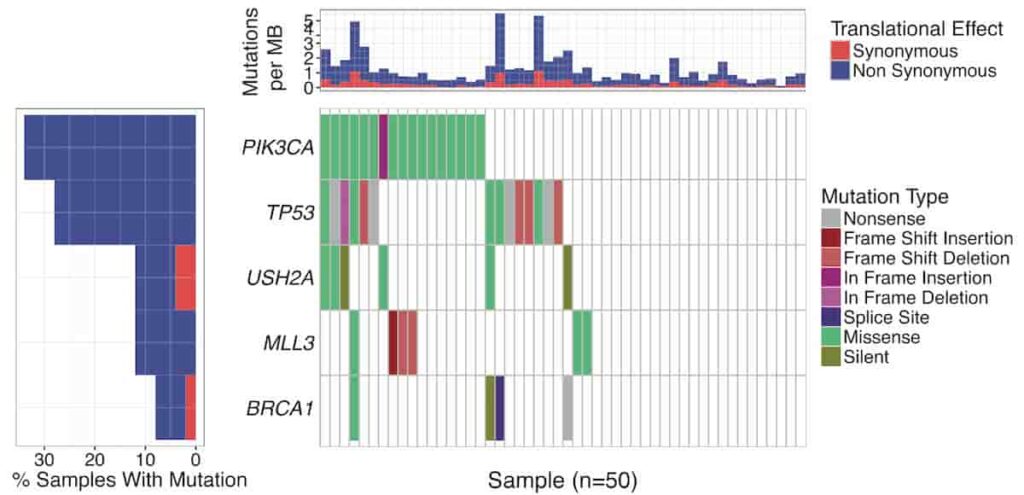
画像出典:Bioinformatics. 2016 Oct 1;32(19):3012-4.
英語のサイト(Griffith Lab)に詳細なやり方が説明されていますので、参考にしながらやってみたいと思います。なお、本記事の最後に書きますが、現在(2024年10月)はGenVisRがアップデートされ、このサイトに書いてある通りのスクリプトではエラーが出てしまうため、以下のCurrent Protocols誌の論文を参考にして少し改良したスクリプトをこれから紹介していきます。
Exploring the Genomic Landscape of Cancer Patient Cohorts with GenVisR(出典:Curr Protoc. 2021 Sep;1(9):e252. doi: 10.1002/cpz1.252.)
準備:GenVisRパッケージのインストールとデータの読み込み
パッケージのインストール
まず、R studioを立ち上げて、パッケージ「GenVisR」をインストールします。
# Install and load GenVisR
# GenVisRをインストール&ロードする
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("GenVisR")
library(GenVisR)
library(data.table)Rのパッケージは、一般的に3箇所(CRAN、Bioconductor、GitHub)のいずれかからダウンロードしますが、GenVisRはBioconductorからダウンロードできます。
ダウンロードに時間がかかるため、wifi環境が整った場所で、時間に余裕がある時にインストールを開始することをおすすめします。
上に書いたスクリプトで、最初の「if…」から3行を実行するとインストールが開始されます。
インストールが終わったら、「library…」でパッケージをロードしましょう。
ちなみに「#」はハッシュと呼ばれ、#を頭につけた行は、スクリプトとして認識せず、メモとして使用するものになります。
データの読み込み
次に、waterfall plotとして図にしたいデータを読み込ませます。
今回は、Ma CX, et al. (2015)で公開されているデータを用いてwaterfall plotを描いてみます(出典:Clin Cancer Res. 2016 Apr 1;22(7):1583-91.)
mutationのデータ、臨床データ、mutation burdenの3種類のデータを読み込みます。
自分の実験データを用いる際には、このデータと同じ形式に自分のデータを加工する必要があります。いずれもtsvとなっていますので、自分でデータを加工する際には、excelで作成したのちに、「タブ区切りテキスト(.txt)」で保存すると、以下と同じようにread.delim関数で読み込むことができます。
# Load relevant data from the manuscript
#遺伝子情報のデータを読み込む
mutationData <- read.delim("http://genomedata.org/gen-viz-workshop/GenVisR/BKM120_Mutation_Data.tsv")
clinicalData <- read.delim("http://genomedata.org/gen-viz-workshop/GenVisR/BKM120_Clinical.tsv")
mutationBurden <- read.delim("http://genomedata.org/gen-viz-workshop/GenVisR/BKM120_MutationBurden.tsv")
データの確認
# データを確認する
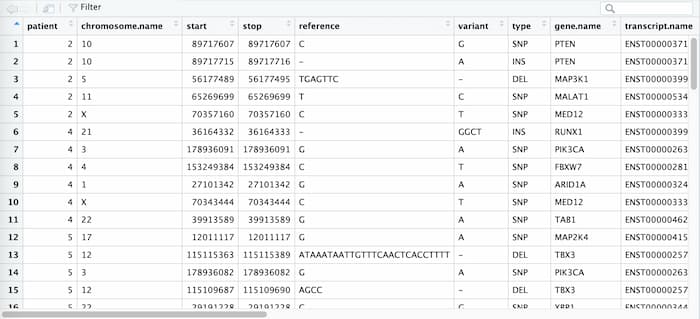
view(mutationData)
mutationDataは、以下のような形式として読み込まれているのが確認できます(R studioを使用した場合には以下のように表示されます)。
Waterfall plotを描く
Waterfall関数で図を描く
次に、waterfall plotをまず描いてみます。詳細なカスタマイズは次の項ですることとして、以下のスクリプトを書いて実行します。
# Reformat the mutation data for Waterfall()
# Waterfall関数で使用できるデータの形式に合わせる
mutationData <- mutationData[,c("patient", "gene.name", "trv.type", "amino.acid.change")]
colnames(mutationData) <- c("sample", "gene", "mutation", "amino.acid.change")
# Create a vector to save mutation priority order for plotting
# Mutationの種類と順番を設定して色を当てる
myHierarchy <- data.table("mutation"=
c("nonsense", "frame_shift_del", "frame_shift_ins", "in_frame_del",
"splice_site_del", "splice_site", "missense","splice_region", "rna"),
color=c("#FF0000", "#00A08A", "#F2AD00", "#F98400",
"#5BBCD6", "#046C9A", "#D69C4E", "#000000", "#446455"))
# Create and sava an initial plot
# 最初のfigureを作成して保存する
plotData <- Waterfall(mutationData, mutationHierarchy = myHierarchy)
pdf(file="Figure_1.pdf", height=8, width=12)
drawPlot(plotData)
dev.off()スクリプトの内容を一つずつ見ていきましょう。
まず、読み込んだデータ「mutationData」から、4つの列(”patient”, “gene.name”, “trv.type”, “amino.acid.change”)だけ、取り出します。そして、4つの列に新しい列名を当てます(”sample”, “gene”, “mutation”, “amino.acid.change”)。
次に、Mutationの種類と順番を指定し、それぞれのMutationに色を当てます。自分の実験データでfigureを作成する際には、自分のデータに合うようにここを修正します。ここが一番エラーが起きる可能性があるので、次の「トラブルシューティング」の項目で、解決策をいくつか紹介します。
最後に、Waterfall関数を使って、figureを作成します。作成されたfigureを一度「plotData」に入れ、「pdf(file=”Figure_1.pdf …」のスクリプトでPDFに書き出します。
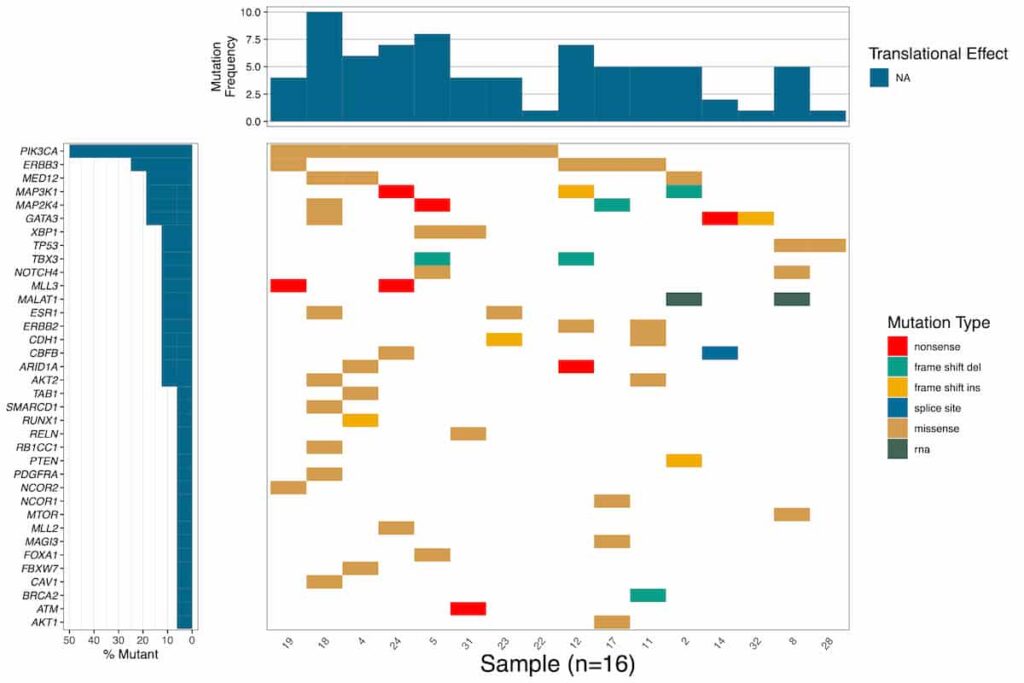
これで表が完成され、PDFとして保存されました。
どこに保存されたか分からない場合は、スクリプトに「getwd()」と打って、自分が今どこのフォルダにいるかを確認しましょう。
トラブルシューティング
自分の実験データで行う際は、「Mutationの種類と順番を指定し、それぞれのMutationに色を当てる」ところで躓きやすいため、いくつか解決策を書いておきます。
警告メッセージ:
setMutationHierarchy(object, mutationHierarchy = mutationHierarchy, で:
The following mutations were found in the input however were not specified in the mutationHierarchy! NA adding these in as least important and assigning random colors!
このようなメッセージが出ることがあります。他にもエラーが出る原因は3つほど考えられますので、ここにまとめておきましょう。
エラー1:mutationの種類を全て書き出していない
mutationData の mutation 列に、ある全てのmutationの種類を書き出して、スクリプトのmyHierarchy の部分に記載する必要があります(今回の例では、”mutation”= c(“nonsense”, “frame_shift_del”, “frame_shift_ins”, “in_frame_del”, “splice_site_del”, “splice_site”, “missense”,”splice_region”, “rna”))。
エラーが出たら、myHierarchy に含まれていないmutaionの種類があるか確認してください。
元々のデータで一つずつ探してもいいですし、元データをExcelで開いてフィルター機能を使って見ることも可能ですが、Rで解決する方法もあります。
次のコードを使うと、mutationData のmutation内に存在し、myHierarchy に書き出していないmutaionの種類を確認することができます。
# variant_classに含まれるバリアントを確認
unique_variants <- unique(mutationData$variant_class)
# Hierarchyに含まれていないバリアントを確認
setdiff(unique_variants, myHierarchy$mutation)エラー2:色の指定が足りない
myHierarchy にmutaionの種類を全て書き出したら、それぞれに対応する色を指定する必要があります(今回の例では、color=c(“#FF0000”, “#00A08A”, “#F2AD00”, “#F98400”, “#5BBCD6”, “#046C9A”, “#D69C4E”, “#000000”, “#446455”)。
mutaionの種類の数と、色の数が一致しているか確認してみてください。
エラー3:データにNAが含まれる
元々のデータで、mutaionの種類の情報が無い部分に「NA」を記載してあると、データを読み込んだ際に、「NA」という文字ではなく、「情報がないよ、という意味の”NA”」として認識されます。R studioでデータを見てみると、以下の通り、灰色のNAとして表示されています。
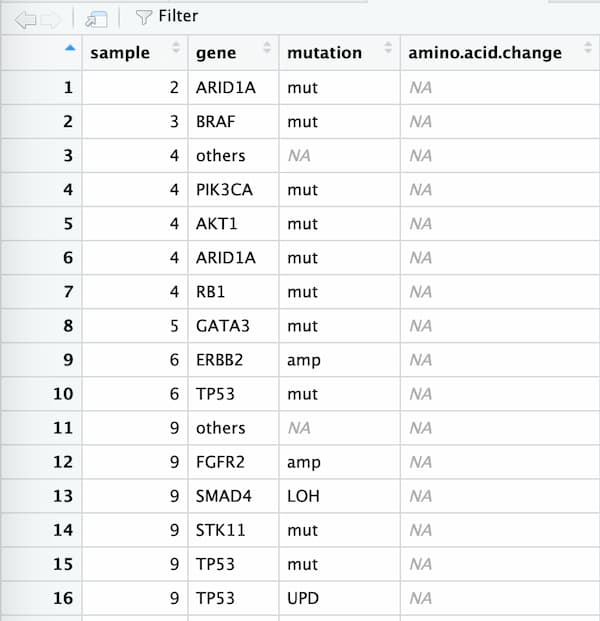
これだと、mutationの種類としては1つとカウントされるのに、myHierarchyでは”NA”と記載してもカウントされないため、エラーが起きてしまいます。
この問題は、1)元データで「NA」を「other」など別の文字列に書き換えるか、2)以下のコードを使ってNAを文字列に変換することで、解決できます。
# NAを文字列に変換
mutationData$mutation[is.na(mutationData$mutation)] <- "NA"Waterfall plotに臨床データを追加する
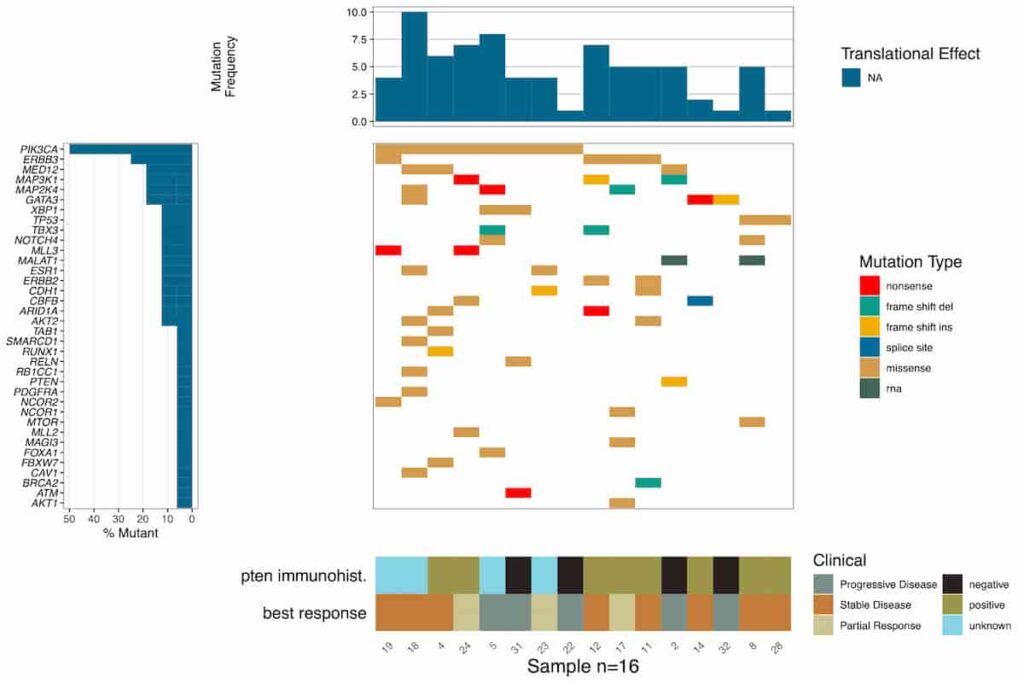
臨床データも追加してみましょう。
myClinical <- clinicalData
myClinical <- myClinical[, c("sample.Number", "Best.response", "PTEN.Immunohist.")]
setnames(myClinical, c("sample", "Best response", "PTEN Immunohist."))
myClinical[,sample := gsub("WU0+","",sample)]
myClinicalColors <- c("Progressive Disease"="#798E87", "Stable Disease"="#C27D38", "Partial Response"="#CCC591", "negative"="#29211F", "positive"="#9C964A", "unknown"="#85D4E3")
clinicalData <- Clinical(inputData = myClinical, palette = myClinicalColors, legendColumns = 2)
plotData <- Waterfall(mutationData, mutationHierarchy = myHierarchy, clinical = clinicalData)
pdf(file="Figure_4.pdf", height=8, width=12)
drawPlot(plotData)
dev.off()
このような臨床データを含んだfigureを描くことができます。
Waterfall plotの更なるカスタマイズ
Current Protocols誌の論文には、mutation burdenの部分をカスタマイズしたり、パッケージggplot2を用いた更なるカスタマイズが紹介されています。
ここでは紹介しませんが、興味がある方は論文を確認してみてください。
「エラー: 使われていない引数」について
Griffith Labのスクリプトそのままではエラーが起きてしまう理由について書いておきます。
昨年(2023年6月)、Griffith Labのサイトでこのやり方を見つけて実行してみたところ、うまくfigureを書くことができました。スクリプトを書き換えることで、自分の実際の実験データをfigureにすることも可能でした。
今年(2024年9月)、再度同じスクリプトを走らせようとしましたが、以下の部分でエラーが出てしまい、なぜかうまくいきませんでした。
> # Create an initial plot
> Waterfall(mutationData, fileType = “Custom”, variant_class_order=mutation_priority)
Waterfall(mutationData, fileType = “Custom”, variant_class_order = mutation_priority) でエラー:
使われていない引数 (fileType = “Custom”, variant_class_order = mutation_priority)
Rのバージョンを新しくしてみたり、スクリプトのエラーをchatGPTに入れて質問をしてみたり、色々と試してみましたが、解決しません。
最終的に解決した方法は、Rのパッケージのマニュアルを確認することでした。以下のように、必ずRのパッケージにはマニュアルがあります。GenVisRをインストールしたBioconductorのサイトからマニュアルを見ることができます。

マニュアル内で、waterfall関数について探してみると、以下のような記載がありました。
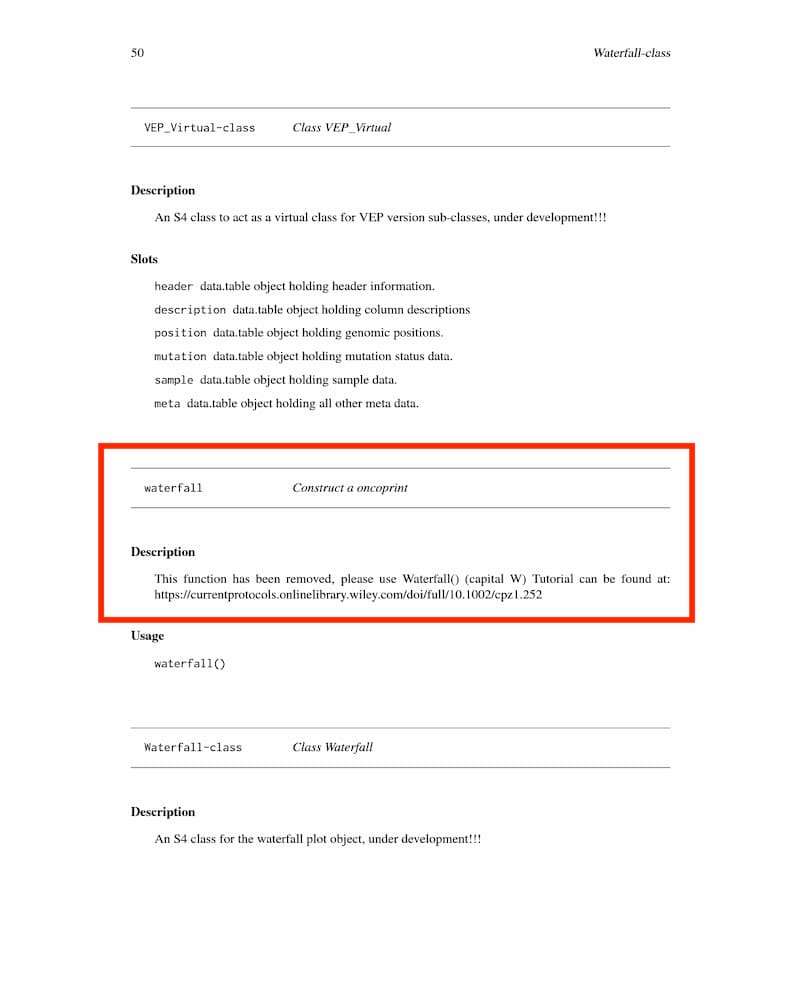
要は、GenVisRのアップデートにより、「waterfall()」という関数は削除された、とのことです。大文字で始まる「Waterfall()」という関数が設定されているので、そちらを使うようにとのこと。その使い方のチュートリアルのリンクが記載されていて、最初に紹介したCurrent Protocols誌の論文のページに飛ぶようになっています。
エラーとして出てきた「使われていない引数」は、「waterfall()という関数は存在しないよ」という意味でした。なお、引数は「ひきすう」と読みます。
パッケージのアップデートで、今までの関数が使えなくなってしまうことは、他にも何度か経験しました。エラーが出た時には、パッケージの最新のマニュアルを見直すことが解決につながることもあると覚えておくと良いでしょう。
さいごに
うまくfigureが書けましたか?
今回は、バイオインフォでよく作成するwaterfall plotの描き方について紹介しました。
Rに使い慣れていない方には、分かりにくい部分も多々あるかと思いますので、コメントをいただければ可能な範囲で適宜改良します。他の記事でも少しずつ詳しく書いていきますね。


コメント